1
Дифференциальный диагноз
Спрашивает: Мария
Пол: Мужской
Возраст: 13 лет
Хронические заболевания: не указаны
Здравствуйте, Сдавали с ребёнком генетический анализ крови на "Эпилептическую панель". У ребёнка с семи лет судороги. Врач эпилептолог посоветовал сдать нам генетический анализ крови. Мы обратились в Геномед и там сдали кровь. На днях нам пришёл результат. Диагноз: Дифференциальный диагноз между синдромом Блоха-Сульцбергера и GEFS+. Подскажите, пожалуйста, что это означает? Заранее спасибо за ответ.
С уважением, Мария.
С уважением, Мария.
Похожие и рекомендуемые вопросы
Расшифруйте результат анализа Расшифруйте, пожалуйста результаты анализа. Ребенку...
Гомозиготная мутация в 3 экзоне гена MCOLN1. что это означает? Меня зовут Марина!...
Патогенная мутация Расшифруйте, пожалуйста результаты анализа. Панель Факоматозы и...
Эпилесия и генетика Ребёнку 7,5 лет, девочка. В 3,5 года дебют эпилепсии, долго не...
Выявлен патогенный вариант c 35delG гена GJB2 в гетерозиготном состоянии Перед планированием...
У ребёнка проблемы с печень Подскажите пожалуйста Методом массового параллельного...
Мутация Гипогонадотропный гипогонадизм Мне в 15 лет поставили диагноз Гипогонадотропный...
Синдром Нунан Подскажите пож. Есть ли у дочки Нунан синдром?
Пункт прейскуранта:...
Анализ ДНК Помогите пожалуйста расшифровать анализ. Врачи нам сказали необходимо дальше...
Муковисцидоз Помогите, пожалуйста, разобраться. Моему сыну 1 год. В роддоме брали...
Патогенный вариант с.3207са в гетерозиготном состоянии У старшего ребёнка (сын) болезнь...
Полное секвенирование Сдали анализ, полное секвенирование, на 22 000 генов. Пришел...
Беременность и мутация в генах По итогам прохождения узи с доплером на 26 неделе беременности...
Генетическая консультация Уважаемый врач! Помогите пожалуйста! Очень нуждаюсь в Вашей...
Неустановленный вид Ребёнку в прошлом году поставили диагноз сахарный диабет 1 типа...
Некласичесеая форма ВДКН Ребёнку 1г10 мес, поставили вакцину первую инфантрикс гекса,...
Расшифруйте пожалуйста результат анализа Расшифруйте, пожалуйста результаты анализа....
Точный анализ Помогите пожалуйста. Брат и сестра троюродные, так сложилось что мы...
Гетерозигоная мутация в 3 экзоне гена PDHX, и в 4 -TFG Сдали сыну 4 года генетический...
Дисплазия плода 2 триместр беременности 2 года назад при узи второго триместра у ребеночка...
9 ответов
Не забывайте оценивать ответы врачей, помогите нам улучшить их, задавая дополнительные вопросы по теме этого вопроса.
Также не забывайте благодарить врачей.
Также не забывайте благодарить врачей.

Здравствуйте,
нужно видеть все заключение полностью.
Предоставьте скан или качественное фото.
нужно видеть все заключение полностью.
Предоставьте скан или качественное фото.
0
Платная консультация
Мария 2016-08-09 22:01
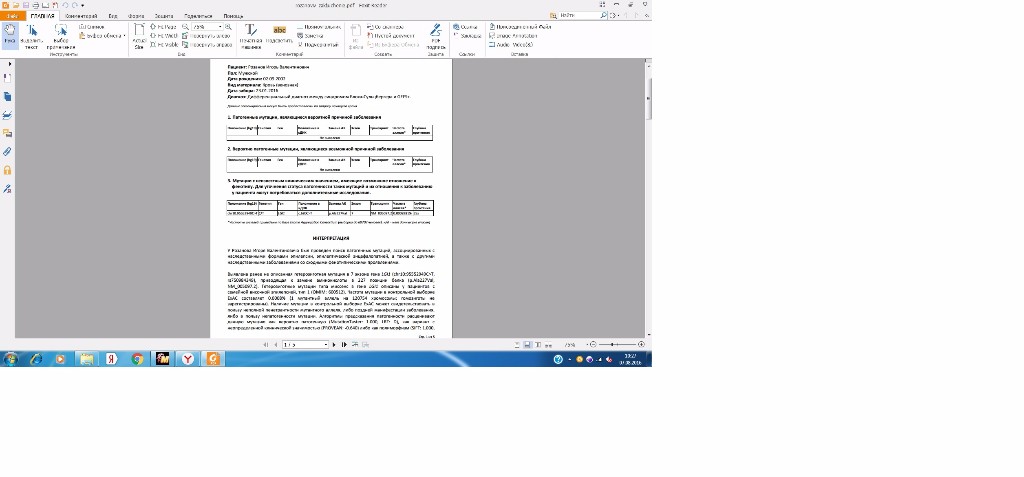
Пациент: Розанов Игорь Валентинович
Пол: Мужской
Дата рождения: 02.09.2002
Вид материала: Кровь (венозная)
Дата забора: 23.01.2016
Диагноз: Дифференциальный диагноз между синдромом Блоха-Сульцбергера и GEFS+.
Данные секвенирования могут быть предоставлены по запросу лечащего врача.
1. Патогенные мутации, являющиеся вероятной причиной заболевания
Положение (hg19) Генотип Ген Положение в
кДНК
Замена АК Экзон Транскрипт Частота
аллеля*
Глубина
прочтения
Не выявлено
2. Вероятно патогенные мутации, являющиеся возможной причиной заболевания
Положение (hg19) Генотип Ген Положение в
кДНК
Замена АК Экзон Транскрипт Частота
аллеля*
Глубина
прочтения
Не выявлено
3. Мутации с неизвестным клиническим значением, имеющие возможное отношение к
фенотипу. Для уточнения статуса патогенности таких мутаций и их отношения к заболеванию
у пациента могут потребоваться дополнительные исследования.
Положение (hg19) Генотип Ген Положение в
кДНК
Замена АК Экзон Транскрипт Частота
аллеля*
Глубина
прочтения
chr10: 95552949CT C/T LGI1 c.680CT p. Ala227Val 7 NM_005097.2 0.0008281% 25x
*Частоты аллелей приведены по базе Exome Aggregation Consortium (выборка до 60702 человек). н/д = нет данных (не описан)
ИНТЕРПРЕТАЦИЯ
У Розанова Игоря Валентиновича был проведен поиск патогенных мутаций, ассоциированных с
наследственными формами эпилепсии, эпилептической энцефалопатией, а также с другими
наследственными заболеваниями со сходными фенотипическими проявлениями.
Выявлена ранее не описанная гетерозиготная мутация в 7 экзоне гена LGI1 (chr10: 95552949CT,
rs750984349), приводящая к замене аминокислоты в 227 позиции белка (p. Ala227Val,
NM_005097.2). Гетерозиготные мутации типа миссенс в гене LGI1 описаны у пациентов с
семейной височной эпилепсией, тип 1 (OMIM: 600512). Частота мутации в контрольной выборке
ExAC составляет 0.0008% (1 мутантный аллель на 120754 хромосомы; гомозиготы не
зарегистрированы). Наличие мутации в контрольной выборке ExAC может свидетельствовать в
пользу неполной пенетрантности мутантного аллеля, либо поздней манифестации заболевания,
либо в пользу непатогенности мутации. Алгоритмы предсказания патогенности расценивают
данную мутацию как вероятно патогенную (MutationTaster: 1.000, LRT: D), как вариант с
неопределенной клинической значимостью (PROVEAN: -0.640) либо как полиморфизм (SIFT: 1.000, Стр. 2 из 5
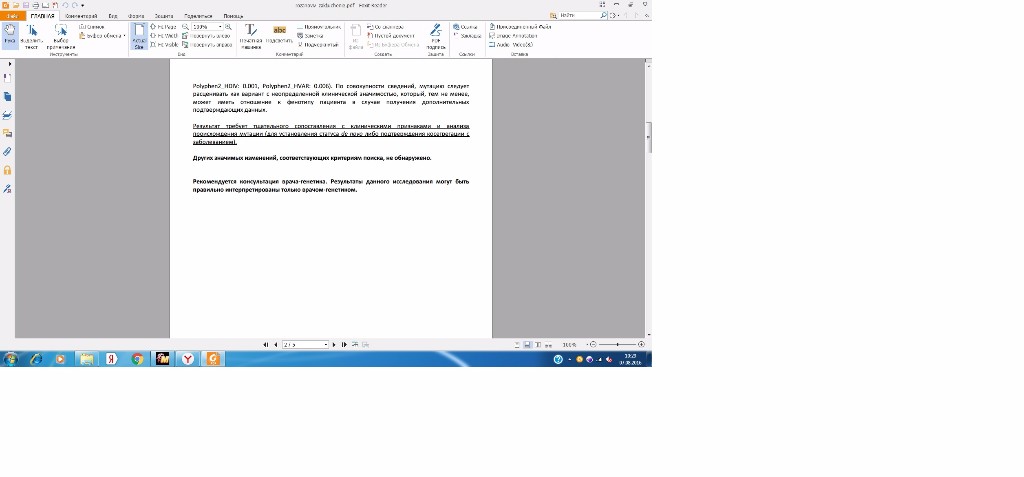
Polyphen2_HDIV: 0.001, Polyphen2_HVAR: 0.006). По совокупности сведений, мутацию следует
расценивать как вариант с неопределенной клинической значимостью, который, тем не менее,
может иметь отношение к фенотипу пациента в случае получения дополнительных
подтверждающих данных.
Результат требует тщательного сопоставления с клиническими признаками и анализа
происхождения мутации (для установления статуса de novo либо подтверждения косегрегации с
заболеванием).
Других значимых изменений, соответствующих критериям поиска, не обнаружено.
Рекомендуется консультация врача-генетика. Результаты данного исследования могут быть
правильно интерпретированы только врачом-генетиком. Стр. 3 из 5
ОПИСАНИЕ ИССЛЕДОВАНИЯ
Анализ ДНК пациента проведен на секвенаторе нового поколения Illumina NextSeq 500 методом
парно-концевого чтения (2x151 п. о.) со средним покрытием не менее 70-100х. Для
пробоподготовки была использована методика селективного захвата участков ДНК, относящихся к
кодирующим областям генов с известным клиническим значением, в том числе AARS, ABCC8,
ABCD1, ACADM, ACADS, ACTB, ACTG1, ACY1, ADAR, ADCK3, ADNP, ADSL, AFG3L2, AGA, AHI1, AIMP1,
AKT3, ALDH4A1, ALDH5A1, ALDH7A1, ALG1, ALG11, ALG12, ALG13, ALG2, ALG3, ALG6, ALG8, ALG9,
AMACR, AMT, ANK3, AP1S2, APOC3, APTX, ARFGEF2, ARG1, ARHGEF9, ARL13B, ARSA, ARSB, ARX,
ASAH1, ASL, ASNS, ASPA, ASPM, ASS1, ATIC, ATN1, ATP1A2, ATP1A3, ATP2A2, ATP6AP2, ATP7A, ATR,
ATRX, B4GALT1, BCKDHA, BCKDHB, BCS1L, BRAF, BRAT1, BTD, BUB1B, C12orf57, C5orf42, CACNA1A,
CACNA1H, CACNB4, CASC5, CASK, CASR, CC2D2A, CCDC88C, CDK5RAP2, CDKL5, CDON, CENPJ, CEP135,
CEP152, CEP290, CEP41, CEP63, CHD2, CHRNA2, CHRNA4, CHRNB2, CLCN2, CLIC2, CLN3, CLN5, CLN6,
CLN8, CNNM2, CNTNAP2, COA5, COG1, COG4, COG5, COG6, COG7, COG8, COL18A1, COL4A1, COL4A2,
COQ2, COQ4, COQ6, COQ9, COX10, COX14, COX15, COX6B1, CPA6, CPS1, CPT1A, CPT2, CREBBP, CSTB,
CTSA, CTSD, CUL4B, DBT, DCX, DDOST, DGUOK, DHCR24, DHCR7, DLD, DLG3, DNAJC5, DNM1, DOCK8,
DOLK, DPAGT1, DPM1, DPM3, DPYD, DYNC1H1, DYRK1A, EARS2, EFHC1, EIF2B1, EIF2B2, EIF2B3, EIF2B4,
EIF2B5, EMX2, EPM2A, ETFA, ETFB, ETFDH, ETHE1, EXOSC3, EZH2, FA2H, FAM126A, FASTKD2, FGD1,
FGFR3, FH, FIG4, FKRP, FKTN, FLNA, FOLR1, FOXG1, FOXRED1, FUCA1, GABRA1, GABRB3, GABRD,
GABRG2, GALC, GALNS, GAMT, GATM, GCDH, GCH1, GCK, GCSH, GFAP, GFM1, GJC2, GLB1, GLDC, GLI2,
GLI3, GLRA1, GLRB, GNE, GNPTAB, GNPTG, GNS, GOSR2, GPC3, GPHN, GRIA3, GRIN1, GRIN2A, GRIN2B,
GRN, GUSB, HADH, HCN1, HDAC8, HEPACAM, HERC2, HEXA, HEXB, HGSNAT, HPD, HSD17B10, HSPD1,
HYAL1, IDS, IDUA, IER3IP1, IFIH1, IL1RAPL1, INS, IQSEC2, ISPD, IVD, KCNA1, KCNJ1, KCNJ10, KCNJ11,
KCNK18, KCNMA1, KCNQ2, KCNQ3, KCTD7, KDM5C, KDM6A, KIAA2022, KIF7, KIRREL3, KMT2D, KRIT1,
L2HGDH, LAMA2, LAMB1, LAMC3, LARGE, LGI1, LIAS, MAN1B1, MAP2K1, MAP2K2, MBD5, MCOLN1,
MCPH1, MECP2, MED12, MED17, MEF2C, MFSD2A, MFSD8, MGAT2, MLC1, MMAA, MMACHC, MOCS1,
MOCS2, MOGS, MPDU1, MPI, MTHFR, MUT, NAGLU, NAGS, NDE1, NDST1, NDUFA1, NDUFA10,
NDUFA11, NDUFA12, NDUFA2, NDUFA9, NDUFAF1, NDUFAF2, NDUFAF3, NDUFAF4, NDUFAF5, NDUFB3,
NDUFB9, NDUFS1, NDUFS2, NDUFS3, NDUFS4, NDUFS6, NDUFS7, NDUFS8, NDUFV1, NDUFV2, NEU1,
NF1, NFIX, NHLRC1, NIPBL, NOTCH3, NPC1, NPC2, NPHP1, NRAS, NRXN1, NSD1, NSUN2, NUBPL, OCLN,
OCRL, OFD1, OPHN1, OTC, PAFAH1B1, PAH, PAK3, PANK2, PC, PCBD1, PCCA, PCCB, PCDH19, PCNT,
PDHA1, PDHX, PDP1, PDSS1, PDSS2, PEX1, PEX10, PEX11B, PEX12, PEX13, PEX14, PEX16, PEX19, PEX2,
PEX26, PEX3, PEX5, PEX6, PEX7, PGK1, PGM1, PHF6, PIGA, PIGN, PIGO, PIGV, PIK3CA, PIK3R2, PLA2G6,
PLCB1, PLP1, PMM2, PNKP, PNPO, POLG, POLR3A, POLR3B, POMGNT1, POMT1, POMT2, PPT1, PQBP1,
PRICKLE1, PRODH, PRPS1, PRRT2, PSAP, PTCH1, PTEN, PTS, QDPR, RAB18, RAB39B, RAB3GAP1,
RAB3GAP2, RAD21, RAI1, RARS2, RBBP8, RELN, RFT1, RNASEH2A, RNASEH2B, RNASEH2C, RNASET2,
ROGDI, RPGRIP1L, RPS6KA3, RRM2B, SAMHD1, SCARB2, SCN1A, SCN1B, SCN2A, SCN8A, SCN9A, SDHA,
SDHAF1, SEPSECS, SERPINI1, SETBP1, SGCE, SGSH, SHH, SIX3, SLC17A5, SLC19A3, SLC1A3, SLC25A15,
SLC25A19, SLC25A20, SLC25A22, SLC2A1, SLC33A1, SLC35A1, SLC35C1, SLC46A1, SLC6A5, SLC6A8,
SLC9A6, SMARCA2, SMARCB1, SMC1A, SMC3, SMPD1, SMS, SNAP29, SOX10, SPTAN1, SRD5A3, SRPX2,
ST3GAL3, ST3GAL5, STIL, STXBP1, SUCLA2, SUMF1, SUOX, SURF1, SYN1, SYNGAP1, SYP, TACO1,
TBC1D24, TBCE, TBP, TBX1, TCF4, TCTN1, TCTN2, TGIF1, TMEM138, TMEM165, TMEM216, TMEM237,
TMEM67, TMEM70, TPP1, TRAPPC9, TREX1, TSC1, TSC2, TSEN2, TSEN34, TSEN54, TSFM, TTC21B,
TUBA1A, TUBA8, TUBB2B, TUBB3, TUSC3, UBE2A, UBE3A, VLDLR, VPS13A, VPS13B, VRK1, WDR45,
WDR62, WFS1, WWOX, ZEB2, ZIC2, ZNF335.
Обработка данных секвенирования проведена с использованием автоматизированного
алгоритма, включающего выравнивание прочтений на референсную последовательность генома
человека (hg19), постпроцессинг выравнивания, выявление вариантов и фильтрацию вариантов
по качеству, а также аннотацию выявленных вариантов по всем известным транскриптам каждого
гена из базы RefSeq с применением ряда методов предсказания патогенности замен (SIFT, Стр. 4 из 5
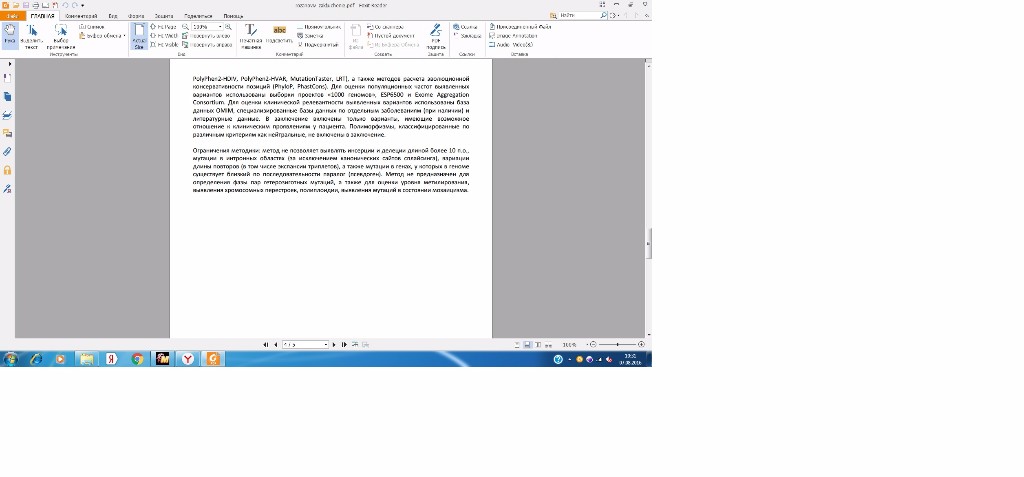
PolyPhen2-HDIV, PolyPhen2-HVAR, MutationTaster, LRT), а также методов расчета эволюционной
консервативности позиций (PhyloP, PhastCons). Для оценки популяционных частот выявленных
вариантов использованы выборки проектов «1000 геномов», ESP6500 и Exome Aggregation
Consortium. Для оценки клинической релевантности выявленных вариантов использованы база
данных OMIM, специализированные базы данных по отдельным заболеваниям (при наличии) и
литературные данные. В заключение включены только варианты, имеющие возможное
отношение к клиническим проявлениям у пациента. Полиморфизмы, классифицированные по
различным критериям как нейтральные, не включены в заключение.
Ограничения методики: метод не позволяет выявлять инсерции и делеции длиной более 10 п. о.,
мутации в интронных областях (за исключением канонических сайтов сплайсинга), вариации
длины повторов (в том числе экспансии триплетов), а также мутации в генах, у которых в геноме
существует близкий по последовательности паралог (псевдоген). Метод не предназначен для
определения фазы пар гетерозиготных мутаций, а также для оценки уровня метилирования,
выявления хромосомных перестроек, полиплоидии, выявления мутаций в состоянии мозаицизма.
Пол: Мужской
Дата рождения: 02.09.2002
Вид материала: Кровь (венозная)
Дата забора: 23.01.2016
Диагноз: Дифференциальный диагноз между синдромом Блоха-Сульцбергера и GEFS+.
Данные секвенирования могут быть предоставлены по запросу лечащего врача.
1. Патогенные мутации, являющиеся вероятной причиной заболевания
Положение (hg19) Генотип Ген Положение в
кДНК
Замена АК Экзон Транскрипт Частота
аллеля*
Глубина
прочтения
Не выявлено
2. Вероятно патогенные мутации, являющиеся возможной причиной заболевания
Положение (hg19) Генотип Ген Положение в
кДНК
Замена АК Экзон Транскрипт Частота
аллеля*
Глубина
прочтения
Не выявлено
3. Мутации с неизвестным клиническим значением, имеющие возможное отношение к
фенотипу. Для уточнения статуса патогенности таких мутаций и их отношения к заболеванию
у пациента могут потребоваться дополнительные исследования.
Положение (hg19) Генотип Ген Положение в
кДНК
Замена АК Экзон Транскрипт Частота
аллеля*
Глубина
прочтения
chr10: 95552949CT C/T LGI1 c.680CT p. Ala227Val 7 NM_005097.2 0.0008281% 25x
*Частоты аллелей приведены по базе Exome Aggregation Consortium (выборка до 60702 человек). н/д = нет данных (не описан)
ИНТЕРПРЕТАЦИЯ
У Розанова Игоря Валентиновича был проведен поиск патогенных мутаций, ассоциированных с
наследственными формами эпилепсии, эпилептической энцефалопатией, а также с другими
наследственными заболеваниями со сходными фенотипическими проявлениями.
Выявлена ранее не описанная гетерозиготная мутация в 7 экзоне гена LGI1 (chr10: 95552949CT,
rs750984349), приводящая к замене аминокислоты в 227 позиции белка (p. Ala227Val,
NM_005097.2). Гетерозиготные мутации типа миссенс в гене LGI1 описаны у пациентов с
семейной височной эпилепсией, тип 1 (OMIM: 600512). Частота мутации в контрольной выборке
ExAC составляет 0.0008% (1 мутантный аллель на 120754 хромосомы; гомозиготы не
зарегистрированы). Наличие мутации в контрольной выборке ExAC может свидетельствовать в
пользу неполной пенетрантности мутантного аллеля, либо поздней манифестации заболевания,
либо в пользу непатогенности мутации. Алгоритмы предсказания патогенности расценивают
данную мутацию как вероятно патогенную (MutationTaster: 1.000, LRT: D), как вариант с
неопределенной клинической значимостью (PROVEAN: -0.640) либо как полиморфизм (SIFT: 1.000, Стр. 2 из 5
Polyphen2_HDIV: 0.001, Polyphen2_HVAR: 0.006). По совокупности сведений, мутацию следует
расценивать как вариант с неопределенной клинической значимостью, который, тем не менее,
может иметь отношение к фенотипу пациента в случае получения дополнительных
подтверждающих данных.
Результат требует тщательного сопоставления с клиническими признаками и анализа
происхождения мутации (для установления статуса de novo либо подтверждения косегрегации с
заболеванием).
Других значимых изменений, соответствующих критериям поиска, не обнаружено.
Рекомендуется консультация врача-генетика. Результаты данного исследования могут быть
правильно интерпретированы только врачом-генетиком. Стр. 3 из 5
ОПИСАНИЕ ИССЛЕДОВАНИЯ
Анализ ДНК пациента проведен на секвенаторе нового поколения Illumina NextSeq 500 методом
парно-концевого чтения (2x151 п. о.) со средним покрытием не менее 70-100х. Для
пробоподготовки была использована методика селективного захвата участков ДНК, относящихся к
кодирующим областям генов с известным клиническим значением, в том числе AARS, ABCC8,
ABCD1, ACADM, ACADS, ACTB, ACTG1, ACY1, ADAR, ADCK3, ADNP, ADSL, AFG3L2, AGA, AHI1, AIMP1,
AKT3, ALDH4A1, ALDH5A1, ALDH7A1, ALG1, ALG11, ALG12, ALG13, ALG2, ALG3, ALG6, ALG8, ALG9,
AMACR, AMT, ANK3, AP1S2, APOC3, APTX, ARFGEF2, ARG1, ARHGEF9, ARL13B, ARSA, ARSB, ARX,
ASAH1, ASL, ASNS, ASPA, ASPM, ASS1, ATIC, ATN1, ATP1A2, ATP1A3, ATP2A2, ATP6AP2, ATP7A, ATR,
ATRX, B4GALT1, BCKDHA, BCKDHB, BCS1L, BRAF, BRAT1, BTD, BUB1B, C12orf57, C5orf42, CACNA1A,
CACNA1H, CACNB4, CASC5, CASK, CASR, CC2D2A, CCDC88C, CDK5RAP2, CDKL5, CDON, CENPJ, CEP135,
CEP152, CEP290, CEP41, CEP63, CHD2, CHRNA2, CHRNA4, CHRNB2, CLCN2, CLIC2, CLN3, CLN5, CLN6,
CLN8, CNNM2, CNTNAP2, COA5, COG1, COG4, COG5, COG6, COG7, COG8, COL18A1, COL4A1, COL4A2,
COQ2, COQ4, COQ6, COQ9, COX10, COX14, COX15, COX6B1, CPA6, CPS1, CPT1A, CPT2, CREBBP, CSTB,
CTSA, CTSD, CUL4B, DBT, DCX, DDOST, DGUOK, DHCR24, DHCR7, DLD, DLG3, DNAJC5, DNM1, DOCK8,
DOLK, DPAGT1, DPM1, DPM3, DPYD, DYNC1H1, DYRK1A, EARS2, EFHC1, EIF2B1, EIF2B2, EIF2B3, EIF2B4,
EIF2B5, EMX2, EPM2A, ETFA, ETFB, ETFDH, ETHE1, EXOSC3, EZH2, FA2H, FAM126A, FASTKD2, FGD1,
FGFR3, FH, FIG4, FKRP, FKTN, FLNA, FOLR1, FOXG1, FOXRED1, FUCA1, GABRA1, GABRB3, GABRD,
GABRG2, GALC, GALNS, GAMT, GATM, GCDH, GCH1, GCK, GCSH, GFAP, GFM1, GJC2, GLB1, GLDC, GLI2,
GLI3, GLRA1, GLRB, GNE, GNPTAB, GNPTG, GNS, GOSR2, GPC3, GPHN, GRIA3, GRIN1, GRIN2A, GRIN2B,
GRN, GUSB, HADH, HCN1, HDAC8, HEPACAM, HERC2, HEXA, HEXB, HGSNAT, HPD, HSD17B10, HSPD1,
HYAL1, IDS, IDUA, IER3IP1, IFIH1, IL1RAPL1, INS, IQSEC2, ISPD, IVD, KCNA1, KCNJ1, KCNJ10, KCNJ11,
KCNK18, KCNMA1, KCNQ2, KCNQ3, KCTD7, KDM5C, KDM6A, KIAA2022, KIF7, KIRREL3, KMT2D, KRIT1,
L2HGDH, LAMA2, LAMB1, LAMC3, LARGE, LGI1, LIAS, MAN1B1, MAP2K1, MAP2K2, MBD5, MCOLN1,
MCPH1, MECP2, MED12, MED17, MEF2C, MFSD2A, MFSD8, MGAT2, MLC1, MMAA, MMACHC, MOCS1,
MOCS2, MOGS, MPDU1, MPI, MTHFR, MUT, NAGLU, NAGS, NDE1, NDST1, NDUFA1, NDUFA10,
NDUFA11, NDUFA12, NDUFA2, NDUFA9, NDUFAF1, NDUFAF2, NDUFAF3, NDUFAF4, NDUFAF5, NDUFB3,
NDUFB9, NDUFS1, NDUFS2, NDUFS3, NDUFS4, NDUFS6, NDUFS7, NDUFS8, NDUFV1, NDUFV2, NEU1,
NF1, NFIX, NHLRC1, NIPBL, NOTCH3, NPC1, NPC2, NPHP1, NRAS, NRXN1, NSD1, NSUN2, NUBPL, OCLN,
OCRL, OFD1, OPHN1, OTC, PAFAH1B1, PAH, PAK3, PANK2, PC, PCBD1, PCCA, PCCB, PCDH19, PCNT,
PDHA1, PDHX, PDP1, PDSS1, PDSS2, PEX1, PEX10, PEX11B, PEX12, PEX13, PEX14, PEX16, PEX19, PEX2,
PEX26, PEX3, PEX5, PEX6, PEX7, PGK1, PGM1, PHF6, PIGA, PIGN, PIGO, PIGV, PIK3CA, PIK3R2, PLA2G6,
PLCB1, PLP1, PMM2, PNKP, PNPO, POLG, POLR3A, POLR3B, POMGNT1, POMT1, POMT2, PPT1, PQBP1,
PRICKLE1, PRODH, PRPS1, PRRT2, PSAP, PTCH1, PTEN, PTS, QDPR, RAB18, RAB39B, RAB3GAP1,
RAB3GAP2, RAD21, RAI1, RARS2, RBBP8, RELN, RFT1, RNASEH2A, RNASEH2B, RNASEH2C, RNASET2,
ROGDI, RPGRIP1L, RPS6KA3, RRM2B, SAMHD1, SCARB2, SCN1A, SCN1B, SCN2A, SCN8A, SCN9A, SDHA,
SDHAF1, SEPSECS, SERPINI1, SETBP1, SGCE, SGSH, SHH, SIX3, SLC17A5, SLC19A3, SLC1A3, SLC25A15,
SLC25A19, SLC25A20, SLC25A22, SLC2A1, SLC33A1, SLC35A1, SLC35C1, SLC46A1, SLC6A5, SLC6A8,
SLC9A6, SMARCA2, SMARCB1, SMC1A, SMC3, SMPD1, SMS, SNAP29, SOX10, SPTAN1, SRD5A3, SRPX2,
ST3GAL3, ST3GAL5, STIL, STXBP1, SUCLA2, SUMF1, SUOX, SURF1, SYN1, SYNGAP1, SYP, TACO1,
TBC1D24, TBCE, TBP, TBX1, TCF4, TCTN1, TCTN2, TGIF1, TMEM138, TMEM165, TMEM216, TMEM237,
TMEM67, TMEM70, TPP1, TRAPPC9, TREX1, TSC1, TSC2, TSEN2, TSEN34, TSEN54, TSFM, TTC21B,
TUBA1A, TUBA8, TUBB2B, TUBB3, TUSC3, UBE2A, UBE3A, VLDLR, VPS13A, VPS13B, VRK1, WDR45,
WDR62, WFS1, WWOX, ZEB2, ZIC2, ZNF335.
Обработка данных секвенирования проведена с использованием автоматизированного
алгоритма, включающего выравнивание прочтений на референсную последовательность генома
человека (hg19), постпроцессинг выравнивания, выявление вариантов и фильтрацию вариантов
по качеству, а также аннотацию выявленных вариантов по всем известным транскриптам каждого
гена из базы RefSeq с применением ряда методов предсказания патогенности замен (SIFT, Стр. 4 из 5
PolyPhen2-HDIV, PolyPhen2-HVAR, MutationTaster, LRT), а также методов расчета эволюционной
консервативности позиций (PhyloP, PhastCons). Для оценки популяционных частот выявленных
вариантов использованы выборки проектов «1000 геномов», ESP6500 и Exome Aggregation
Consortium. Для оценки клинической релевантности выявленных вариантов использованы база
данных OMIM, специализированные базы данных по отдельным заболеваниям (при наличии) и
литературные данные. В заключение включены только варианты, имеющие возможное
отношение к клиническим проявлениям у пациента. Полиморфизмы, классифицированные по
различным критериям как нейтральные, не включены в заключение.
Ограничения методики: метод не позволяет выявлять инсерции и делеции длиной более 10 п. о.,
мутации в интронных областях (за исключением канонических сайтов сплайсинга), вариации
длины повторов (в том числе экспансии триплетов), а также мутации в генах, у которых в геноме
существует близкий по последовательности паралог (псевдоген). Метод не предназначен для
определения фазы пар гетерозиготных мутаций, а также для оценки уровня метилирования,
выявления хромосомных перестроек, полиплоидии, выявления мутаций в состоянии мозаицизма.
0
Здравствуйте, Мария,
на сколько я поняла, это Ваш врач эпилептолог сформулировал причину направления - "Диагноз: Дифференциальный диагноз между синдромом Блоха-Сульцбергера и GEFS+"
Это не результат исследования.
Результат это предположение не подтвердил.
Из множества проверенных генов, которые могут участвовать в развитии эпилепсии - ничего точно патогенного не найдено.
Найдена одна замена, которая, возможно не патогенная.
Для возможного уточнения нужно провести еще как минимум один анализ ребенку и обоим родителям.
Но вот на сколько это целесообразно - не уверена.
на сколько я поняла, это Ваш врач эпилептолог сформулировал причину направления - "Диагноз: Дифференциальный диагноз между синдромом Блоха-Сульцбергера и GEFS+"
Это не результат исследования.
Результат это предположение не подтвердил.
Из множества проверенных генов, которые могут участвовать в развитии эпилепсии - ничего точно патогенного не найдено.
Найдена одна замена, которая, возможно не патогенная.
Для возможного уточнения нужно провести еще как минимум один анализ ребенку и обоим родителям.
Но вот на сколько это целесообразно - не уверена.
1
Платная консультация